scvelo.tl.velocity
- scvelo.tl.velocity(data, vkey='velocity', mode='stochastic', fit_offset=False, fit_offset2=False, filter_genes=False, groups=None, groupby=None, groups_for_fit=None, constrain_ratio=None, use_raw=False, use_latent_time=None, perc=None, min_r2=0.01, min_likelihood=0.001, r2_adjusted=None, use_highly_variable=True, diff_kinetics=None, copy=False, **kwargs)
Estimates velocities in a gene-specific manner.
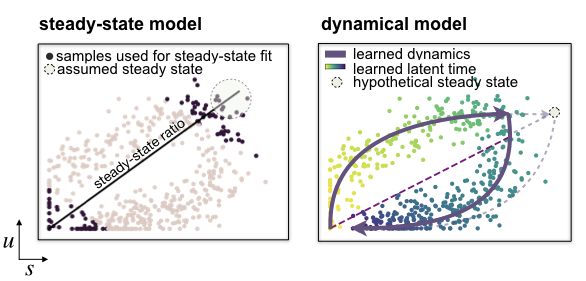
The steady-state model [Manno et al., 2018] determines velocities by quantifying how observations deviate from a presumed steady-state equilibrium ratio of unspliced to spliced mRNA levels. This steady-state ratio is obtained by performing a linear regression restricting the input data to the extreme quantiles. By including second-order moments, the stochastic model [Bergen et al., 2020] exploits not only the balance of unspliced to spliced mRNA levels but also their covariation. By contrast, the likelihood-based dynamical model [Bergen et al., 2020] solves the full splicing kinetics and generalizes RNA velocity estimation to transient systems. It is also capable of capturing non-observed steady states.
- Parameters:
data (
AnnData) – Annotated data matrix.vkey (str (default: ‘velocity’)) – Name under which to refer to the computed velocities for velocity_graph and velocity_embedding.
mode (‘deterministic’, ‘stochastic’ or ‘dynamical’ (default: ‘stochastic’)) – Whether to run the estimation using the steady-state/deterministic, stochastic or dynamical model of transcriptional dynamics. The dynamical model requires to run tl.recover_dynamics first.
fit_offset (bool (default: False)) – Whether to fit with offset for first order moment dynamics.
fit_offset2 (bool, (default: False)) – Whether to fit with offset for second order moment dynamics.
filter_genes (bool (default: True)) – Whether to remove genes that are not used for further velocity analysis.
groups (str, list (default: None)) – Subset of groups, e.g. [‘g1’, ‘g2’, ‘g3’], to which velocity analysis shall be restricted.
groupby (str, list or np.ndarray (default: None)) – Key of observations grouping to consider.
groups_for_fit (str, list or np.ndarray (default: None)) – Subset of groups, e.g. [‘g1’, ‘g2’, ‘g3’], to which steady-state fitting shall be restricted.
constrain_ratio (float or tuple of type float or None: (default: None)) – Bounds for the steady-state ratio.
use_raw (bool (default: False)) – Whether to use raw data for estimation.
use_latent_time (bool`or `None (default: None)) – Whether to use latent time as a regularization for velocity estimation.
perc (float (default: [5, 95])) – Percentile, e.g. 98, for extreme quantile fit.
min_r2 (float (default: 0.01)) – Minimum threshold for coefficient of determination
min_likelihood (float (default: None)) – Minimal likelihood for velocity genes to fit the model on.
r2_adjusted (bool (default: None)) – Whether to compute coefficient of determination on full data fit (adjusted) or extreme quantile fit (None)
use_highly_variable (bool (default: True)) – Whether to use highly variable genes only, stored in .var[‘highly_variable’].
copy (bool (default: False)) – Return a copy instead of writing to adata.
- Returns:
velocity (.layers) – velocity vectors for each individual cell
velocity_genes, velocity_beta, velocity_gamma, velocity_r2 (.var) – parameters